
Introduction — a small story, a number, a question
I remember the salty tang of the clinic coffee and a phone call on a damp Thursday; we had just learned a polymer coating failed in a routine bench test. That moment pushed me to rewrite how I approach a medical device biological evaluation plan—and it changed project timelines for good. In 2017 alone, teams I worked with in Boston saw a three-month delay and roughly $2.4 million in extra testing when cytotoxicity and extractables were underestimated. This is biological evaluation territory: biocompatibility, cytotoxicity, ISO 10993-level thinking (and yes, a dash of stubborn pride). So — how do you keep a device on schedule without cutting corners or inviting a painful recall? I’ll walk you through what I’ve learned over 15+ years in medical device testing and regulatory consulting, and show where teams routinely trip up. Let’s move from that kitchen-table panic to a clearer plan. Next, I’ll dig into the specific flaws I see in standard approaches.

Why standard plans fail: the hidden design flaws and testing gaps
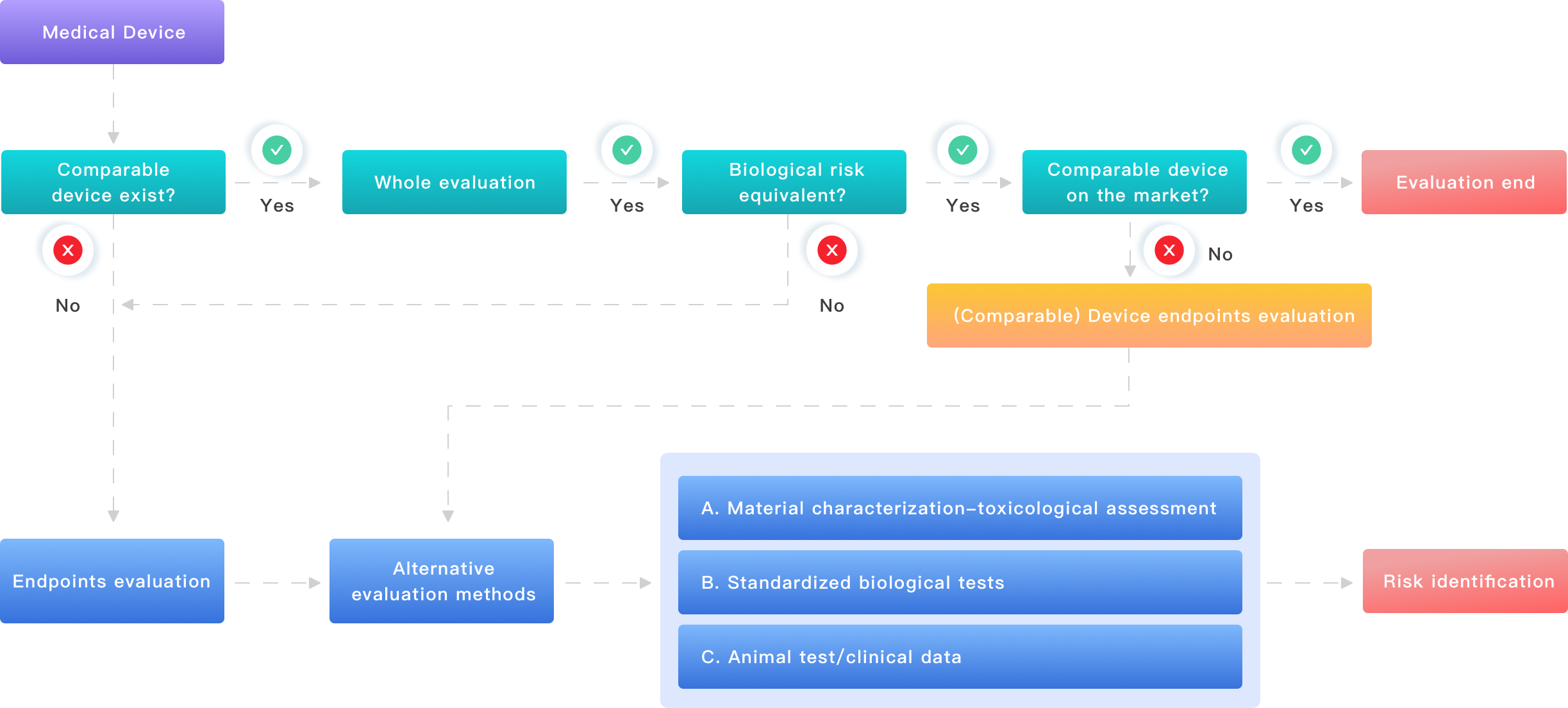
I am blunt about this: most “standard” evaluation plans fail because they are built around hope, not data. I have reviewed dozens of plans for silicone cardiac leads and polyurethane catheters that assumed sterilization validation and extractables profiles would behave the same across suppliers. They did not. The common flaws I see are predictable—poor material traceability, weak risk linkage to biological endpoints, and late-stage testing that triggers redesigns. That translates into measurable pain: a device redesign in Q2 2019 cost my client six weeks of bench time and required repeating endotoxin and cytotoxicity studies. We lost market momentum.
So what goes wrong technically?
First, teams often treat ISO 10993 as a checklist rather than a decision tree. Second, they assume supplier change control is automatic. Third, they schedule extractables and leachables only after final molding and packaging—too late to influence material choice. Those are concrete missteps. I once found an unnoticed adhesive in the finished assembly that altered leachables under sterilization. That discovery forced a repeat of sterilization validation and added cost. Look, I won’t sugarcoat it: these are avoidable if you structure the plan to tie materials, process steps, and biological endpoints together from day one. We document supplier part numbers, lot dates, and sterilization cycles early. That clarity saves months — and money.
Forward-looking fixes: case example and practical measures
When I discuss future outlooks, I prefer examples over slogans. In late 2020, a small OEM in Minneapolis moved from ad hoc testing to a modular evaluation approach. We mapped each component (polymer film, adhesive, coating) to a specific biological endpoint and ran targeted in vitro cytotoxicity and sensitization screens before final assembly. The result: they trimmed three weeks off regulatory submissions and avoided two costly repeat tests. This case shows a principle — plan the biological endpoints, then design tests that answer them directly. It reduces repeat work. — it’s straightforward, though it asks for discipline.
What’s Next?
Looking ahead, I believe labs and R&D teams should pair risk-based planning with early sample triage and parallel testing. That includes quicker in vitro screens and early extractables profiling for new supplier lots. And yes, incorporate biocompatibility testing milestones into procurement calendars. We need to stop treating biocompatibility as a final checkbox and start treating it as an iterative design input. The tools exist—rapid cytotoxicity assays, accelerated extractables techniques, and robust supplier change logs. Use them early. The payoff is faster validation and fewer surprises at submission.
Practical takeaways — three metrics I use to pick a path forward
I’ll close with the three metrics I insist teams track when choosing an evaluation approach. These are concrete and measurable: 1) Time-to-decision for material acceptance (goal: under 30 days from sample receipt), 2) Repeat-test rate after sterilization validation (target: under 10%), and 3) Traceability completeness score (percentage of components with supplier lot ID and material datasheet recorded; aim above 95%). I use these numbers in post-mortems. They tell you where the plan failed and how to fix it. I vividly recall a Saturday morning in March 2016 when a missing lot number cost a client three weeks and $120k; that memory still steers my priorities. I prefer plans that give measurable control, not vague assurances. In the end, smart planning and early, targeted testing change outcomes. For practical support and testing services, I recommend looking into Wuxi AppTec Medical device testing — they have labs and expertise that match these needs.