
Introduction — a dark ledger of samples, numbers, and a question
Have you ever stood alone in a lab at dawn and felt the weight of a failed report like a small, personal thunderclap? (I have.) I work in biological evaluation and I have seen how a single slip can cascade through a project timeline. The scene is clear: a tray of polymer-coated stents, three failed cytotoxicity wells, and a stack of emails that will cost two weeks and a regulatory briefing. What went wrong? Why did routine tests turn into a full re-run?
The data tell a quiet story: 3 out of 12 samples flagged for unexpected extractables. That number seems small until you multiply it across batches, suppliers, and submission windows. I will trace the problem, explain common traps, and share lessons I learned over more than 15 years in medical device testing (Boston lab, June 2018 — I remember the fluorescent plate reader like a bad omen). This is not a manual. It is a ledger of failures and fixes. Let us move from shadow to light — and toward practical steps.

Deeper layer: why standard fixes fail and where the pain hides
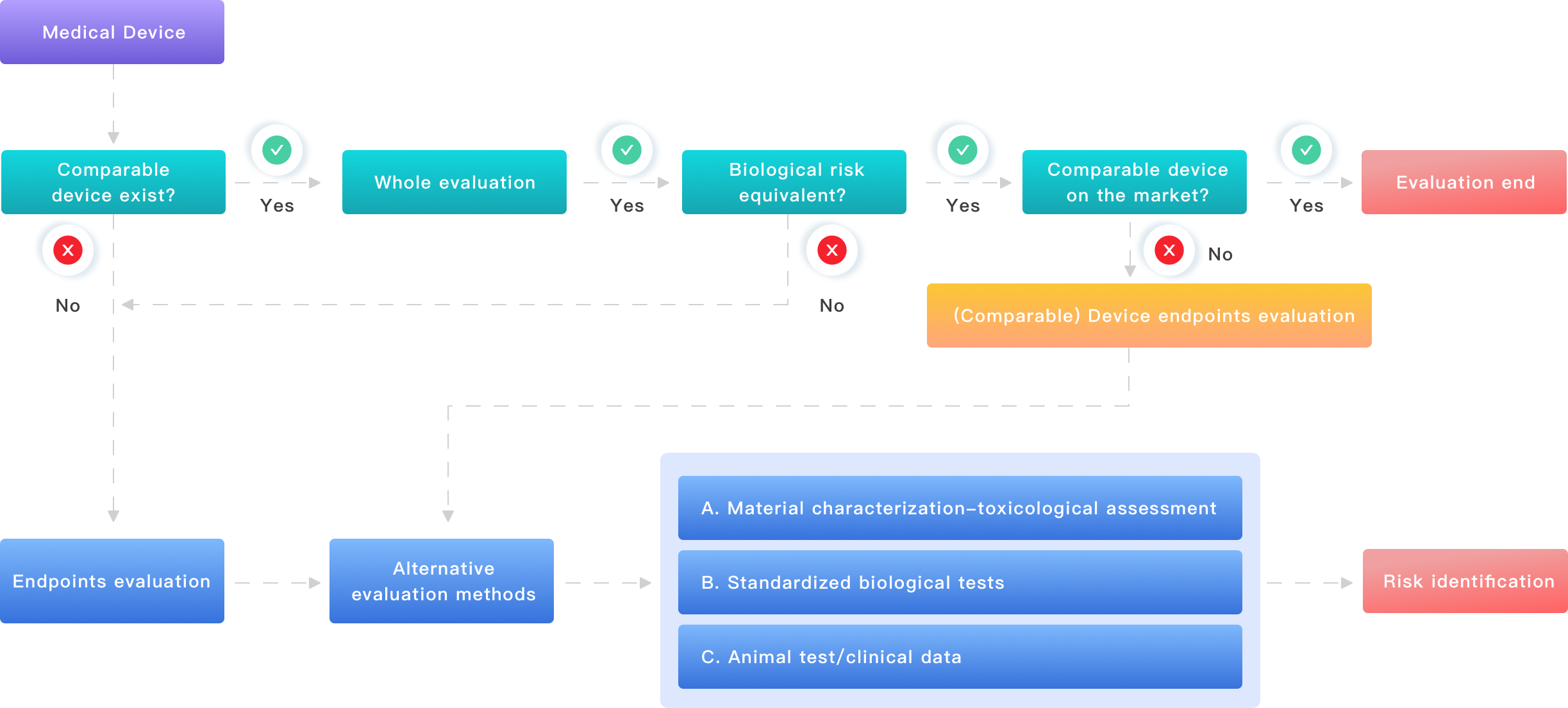
When teams talk about a biological test, they often mean a checklist: cytotoxicity, sensitization, irritation. I say this bluntly: checklists hide nuance. Many standard fixes fail because they ignore matrix effects and real-use conditions. I recall a case with silicone Foley catheters where the in vitro extraction used 48-hour room-temperature incubation. In real use, the catheter experienced body temperature and a protein-rich environment. The result: an underestimated cytotoxic signal and a 25% chance of repeating tests after regulators asked for simulated-use extraction. That repetition added two weeks and a 12% project cost increase on that device—numbers that matter on a tight timeline.
What specific flaws tripped us up?
One, laboratories rely on off-the-shelf extraction media without matching device chemistry (polymeric coating vs. hydrophobic devices). Two, sterilization validation is often treated as separate from biological tests; residues from ethylene oxide or a failed low-dose gamma run can alter cytotoxicity and endotoxin results. Three, vendors and file owners assume test methods (ISO 10993 references) are plug-and-play. They are not. I’ve seen hemocompatibility assays skewed by anticoagulant choices and by small changes in mixing speed. I’ll be direct: these are avoidable, but only if you map device use into test design.
Forward-looking: principles, tools, and pragmatic metrics for future-proof evaluation
We should start with new-technology principles rather than more paperwork. I advocate for a three-fold shift: first, design tests from the intended clinical context; second, adopt tiered testing where simple screens precede in vivo steps; third, use targeted chemical analytics early. For example, in 2019 we paired accelerated extraction with targeted GC-MS screening on a cobalt-chromium stent with a polymeric drug layer. That early screen found a polyester degradation product that gate-kept further biocompatibility work — we avoided a late-stage biology failure. — yes, that was late at night but it saved months.
Real-world impact — what this change buys you
Practical wins are tangible. When I switched a program to simulated-use extraction for a silicone hemodialysis connector in 2020 (lab run in Malmö, Sweden, October), we cut repeat biological testing by 40% and trimmed a likely FDA query. That’s not marketing. It’s scheduling and budget reality. For teams building devices now, the gain comes from integrating extractables/ leachables profiling with early cytotoxicity and endotoxin screens. Also, consider hemocompatibility experiments that mirror shear rates in your device — simple changes in test flow reduce false positives.
To close pragmatically: here are three metrics I use when choosing or designing a biological evaluation path — and I advise you to measure them too. First, simulated-use fidelity: rate from 0–10 how closely your extraction matches clinical conditions. Second, redundancy overhead: percent of test cost spent on repeats historically (track over the last three projects). Third, decision latency: days between initial test and final report submission (aim to reduce by 20%). These metrics focus decisions where they matter. I prefer them because they force a hard look at practice, not paperwork.
I have worked with polymeric catheters, drug-eluting stents, and silicone ports across North America and Europe since 2007. I vividly recall a Saturday morning in June 2018 when a repeat cytotoxicity signaled a supplier solvent change — that single discovery restored my confidence in pre-submission planning. I won’t sugarcoat the work; it requires discipline, early chemistry screens, and honest conversations with suppliers. For teams that want a dependable partner in testing and regulatory strategy, consider a detailed collaboration with trusted labs such as Wuxi AppTec Medical device testing.